
, لديك: 378 مساهمة .
آخر زيارة لك كانت في : الخميس يناير 01, 1970 .
|
|
| | Alkenes...... |  |
| | | كاتب الموضوع | رسالة |
|---|
ابــن الاسلام
إدارة المنتدى


الجنس :   السٌّمعَة : 101 السٌّمعَة : 101  الْمَشِارَكِات : 12744 الْمَشِارَكِات : 12744  النقاط/ : 22644 النقاط/ : 22644  العـمــر : 34 الدولة : العـمــر : 34 الدولة :  المتصفح : المتصفح : 
| موضوع: Alkenes......  الثلاثاء نوفمبر 09, 2010 11:38 am الثلاثاء نوفمبر 09, 2010 11:38 am | |
| Addition Reactions of Alkenes
The most common chemical transformation of a carbon-carbon double bond is the addition reaction.A large number of reagents, both inorganic and organic, have been foundto add to this functional group, and in this section we shall reviewmany of these reactions. A majority of these reactions are exothermic,due to the fact that the C-C pi-bond is relatively weak (ca. 63kcal/mole) relative to the sigma-bonds formed to the atoms or groups ofthe reagent. Remember, the bond energies of a molecule are the energiesrequired to break (homolytically) all the covalent bonds in themolecule. Consequently, if the bond energies of the product moleculesare greater than the bond energies of the reactants, the reaction willbe exothermic. The following calculations for the addition of H-Br aretypical. Note that by convention exothermic reactions have a negative heat of reaction.
[center] 1. Addition of Strong Brønsted Acids As illustrated by the preceding general equation, strongBrønsted acids such as HCl, HBr, HI & H2SO4, rapidly addto the C=C functional group of alkenes to give products in which newcovalent bonds are formed to hydrogen and to the conjugate base of theacid. Using the above equation as a guide, write the addition productsexpected on reacting each of these reagents with cyclohexene.  Weak Brønsted acids such as water (pKa = 15.7) and acetic acid(pKa = 4.75) do not normally add to alkenes. However, the addition of astrong acid serves to catalyze the addition of water, and in this wayalcohols may be prepared from alkenes. For example, if sulfuric acid isdissolved in water it is completely ionized to the hydronium ion,H3O(+), and this strongly acidic (pKa = -1.74) species effectshydration of ethene and other alkenes. CH2=CH2 + H3O(+) ——> HCH2–CH2 OH + H(+) The importance of choosing an appropriate solvent for these additionreactions should now be clear. If the addition of HCl, HBr or HI isdesired, water and alcohols should not be used. These strong acids willionize in such solvents to give ROH2(+) and the nucleophilic oxygen ofthe solvent will compete with the halide anions in the final step,giving alcohol and ether products. By using inert solvents such ashexane, benzene and methylene chloride, these competing solventadditions are avoided. Because these additions proceed by way of polaror ionic intermediates, the rate of reaction is greater in polarsolvents, such as nitromethane and acetonitrile, than in non-polarsolvents, such as cyclohexane and carbon tetrachloride. Regioselectivity and the Markovnikov Rule
Only one product is possible from the addition of these strongacids to symmetrical alkenes such as ethene and cyclohexene. However,if the double bond carbon atoms are not structurally *****alent, as inmolecules of 1-butene, 2-methyl-2-butene and 1-methylcyclohexene, thereagent conceivably may add in two different ways. This is shown for2-methyl-2-butene in the following equation. (CH3)2C=CHCH3 + H-Cl (CH3)2C H–CH ClCH3or(CH3)2C Cl–CH HCH3 2-methyl-2-butene 2-chloro-3-methylbutane 2-chloro-2-methylbutane When addition reactions to such unsymmetrical alkenes are carried out,we find that one of the two possible constitutionally isomeric productsis formed preferentially. Selectivity of this sort is termed regioselectivity.In the above example, 2-chloro-2-methylbutane is nearly the exclusiveproduct. Similarly, 1-butene forms 2-bromobutane as the predominantproduct on treatment with HBr.  After studying many addition reactions of this kind, the Russian chemist Vladimir Markovnikov noticed a trend in the structure of the favored addition product. He formulated this trend as an empirical rule we now call The Markovnikov Rule:When a Brønsted acid, HX, adds to an unsymmetricallysubstituted double bond, the acidic hydrogen of the acid bonds to thatcarbon of the double bond that has the greater number of hydrogen atomsalready attached to it.In more homelier vernacular this rule may be restated as, " Them that has gits." It is a helpful exercise to predict the favored product in examples such as those shown below:  Empirical rules like the Markovnikov Rule are useful aids forremembering and predicting experimental results. Indeed, empiricalrules are often the first step toward practical mastery of a subject,but they seldom constitute true understanding. The Markovnikov Rule,for example, suggests there are common and important principles at workin these addition reactions, but it does not tell us what they are. Thenext step in achieving an understanding of this reaction must be toconstruct a rational mechanistic model that can be tested by experiment.  All the reagents discussed here are strong Brønsted acids so,as a first step, it seems sensible to find a base with which the acidcan react. Since we know that these acids do not react with alkanes, itmust be the pi-electrons of the alkene double bond that serve as thebase. As shown in the diagram on the right, the pi-orbital extends intothe space immediately above and below the plane of the double bond, andthe electrons occupying this orbital may be attracted to the proton ofa Brønsted acid. The resulting acid-base equilibrium generatesa carbocation intermediate (the conjugate acid of the alkene) whichthen combines rapidly with the anionic conjugate base of theBrønsted acid. This two-step mechanism is illustrated for thereaction of ethene with hydrogen chloride by the following equations. First Step: H2C=CH2 + HCl  H HH2C–CH2(+) + Cl(–) Second Step: HH2C–CH2(+) + Cl(–) HH2C–CH2 Cl  An energy diagram for this two-step addition mechanism is shown to theleft. From this diagram we see that the slow or rate-determining step(the first step) is also the product determining step (the anion willnecessarily bond to the carbocation site). Electron donating doublebond substituents increase the reactivity of an alkene, as evidenced bythe increased rate of hydration of 2-methylpropene (two alkyl groups)compared with 1-butene (one alkyl group). Evidently, alkyl substituentsact to increase the rate of addition by lowering the activation energy,ΔE‡1 of the rate determining step, and it is here we should look for arationalization of Markovnikov's rule. As expected, electron withdrawing substituents, such as fluorine orchlorine, reduce the reactivity of an alkene to addition by acids(vinyl chloride is less reactive than ethene).  Energy Energy higher begin lower George Hammond formulated a useful principle that relates thenature of a transition state to its ******** on the reaction path. This Hammond Postulate states that atransition state will be structurally and energetically similar to thespecies (reactant, intermediate or product) nearest to it on thereaction path. In strongly exothermic reactions the transitionstate will resemble the reactant species. In strongly endothermicconversions, such as that shown to the right, the transition state willresemble the high-energy intermediate or product, and will track theenergy of this intermediate if it changes. This change in transitionstate energy and activation energy as the stability of the intermediatechanges may be observed by clicking the higher or lower buttons to theright of the energy diagram. Three examples may be examined, and thereference curve is changed to gray in the diagrams for higher (magenta)and lower (green) energy intermediates. The carbocationintermediate formed in the first step of the addition reaction nowassumes a key role, in that it directly influences the activationenergy for this step. Independent research shows that the stability ofcarbocations varies with the nature of substituents, in a mannersimilar to that seen for alkyl radicals. The exceptional stability ofallyl and benzyl cations is the result of charge delocalization, andthe stabilizing influence of alkyl substituents, although lesspronounced, has been interpreted in a similar fashion. Carbocation
Stability CH3(+) <CH3CH2(+)< (CH3)2CH(+)≈ CH2=CH-CH2(+)<C6H5CH2(+)≈ (CH3)3C(+) From this information, applying the Hammond Postulate, we arrive at a plausible rationalization of Markovnikov's rule. Whenan unsymmetrically substituted double bond is protonated, we expect themore stable carbocation intermediate to be formed faster than the lessstable alternative, because the activation energy of the path tothe former is the lower of the two possibilities. This is illustratedby the following equation for the addition of hydrogen chloride topropene. Note that the initial acid-base equilibrium leads to api-complex which immediately reorganizes to a sigma-bonded carbocationintermediate. The more stable 2º-carbocation is formedpreferentially, and the conjugate base of the Brønsted acid(chloride anion in the example shown below) then rapidly bonds to thiselectrophilic intermediate to form the final product. The following energy diagram summarizes these features. Note that thepi-complex is not shown, since this rapidly and reversibly formedspecies is common to both possible reaction paths. 2. Rearrangement of Carbocations The formation of carbocations is sometimes accompanied by astructural rearrangement. Such rearrangements take place by a shift ofa neighboring alkyl group or hydrogen, and are favored when therearranged carbocation is more stable than the initial cation. Theaddition of HCl to 3,3-dimethyl-1-butene, for example, leads to anunexpected product, 2-chloro-2,3-dimethylbutane, in somewhat greateryield than 3-chloro-2,2-dimethylbutane, the expected Markovnikovproduct. This surprising result may be explained by a carbocationrearrangement of the initially formed 2º-carbocation to a3º-carbocation by a 1,2-shift of a methyl group. To see thisrearrangement click the " Show Mechanism" button to the right of the equation.  Another factor that may induce rearrangement of carbocationintermediates is strain. The addition of HCl to α-pinene, the majorhydrocarbon component of turpentine, gives the rearranged product,bornyl chloride, in high yield. As shown in the following equation,this rearrangement converts a 3º-carbocation to a2º-carbocation, a transformation that is normally unfavorable.However, the rearrangement also expands a strained four-membered ringto a much less-strained five-membered ring, and this relief of strainprovides a driving force for the rearrangement. A three-dimensionalprojection view of the rearrangement may be seen by clicking the " Other View"button. The atom numbers (colored red) for the pinene structure areretained throughout the rearrangement to help orient the viewer. Thegreen numbers in the final product represent the proper numbering ofthis bicyclic ring system.  The propensity for structural rearrangement shown by certain molecularconstitutions, as illustrated above, serves as a useful probe for theintermediacy of carbocations in a reaction. We shall use this testlater. An extensive and more detailed discussion of cation induced rearrangements may be accessed by Clicking Here. 3. Addition of Lewis Acids (Electrophilic Reagents) The proton is not the only electrophilic species that initiatesaddition reactions to the double bond. Lewis acids like the halogens,boron hydrides and certain transition ****l ions are able to bond tothe alkene pi-electrons, and the resulting complexes rearrange or areattacked by nucleophiles to give addition products. The electrophiliccharacter of the halogens is well known. Although fluorine isuncontrollably reactive, chlorine, bromine and to a lesser degreeiodine react selectively with the double bond of alkenes. The additionof chlorine and bromine to alkenes, as shown in the following generalequation, proceeds by an initial electrophilic attack on thepi-electrons of the double bond. Iodine adds reversibly to doublebonds, but the equilibrium does not normally favor the additionproduct, so it is not a useful preparative method. Dihalo-compounds inwhich the halogens are juxtaposed in the manner shown are called vicinal, from the Latin vicinalis, meaning neighboring. R2C=CR2 + X2 ——> R2C X-CR2 X Other halogen containing reagents which add to double bonds include hypohalous acids, HOX,and sulfenyl chlorides, RSCl. These reagents are unsymmetrical, sotheir addition to unsymmetrical double bonds may in principle takeplace in two ways. In practice, these addition reactions areregioselective, with one of the two possible constitutionally isomericproducts being favored. The electrophilic moiety of these reagents isthe halogen. (CH3)2C=CH2 + HOBr ——> (CH3)2C OH-CH2 Br(CH3)2C=CH2 + C6H5 SCl ——> (CH3)2C Cl-CH2 SC6H5 The regioselectivity of the above reactions may be explained by thesame mechanism we used to rationalize the Markovnikov rule. Thus,bonding of an electrophilic species to the double bond of an alkeneshould result in preferential formation of the more stable (more highlysubstituted) carbocation, and this intermediate should then combinerapidly with a nucleophilic species to produce the addition product.This is illustrated by the following equation. To apply this mechanism we need to determine the electrophilic moiety in each of the reagents.  By using electronegativity differences we can dissect common additionreagents into electrophilic and nucleophilic moieties, as shown on theright. In the case of hypochlorous and hypobromous acids (HOX), theseweak Brønsted acids (pKa's ca. 8) do not react as protondonors; and since oxygen is more electronegative than chlorine orbromine, the electrophile will be a halide cation. The nucleophilicspecies that bonds to the intermediate carbocation is then hydroxideion, or more likely water (the usual solvent for these reagents), andthe products are called halohydrins. Sulfenyl chlorides add in theopposite manner because the electrophile is a sulfur cation, RS(+),whereas the nucleophilic moiety is chloride anion (chlorine is moreelectronegative than sulfur). If you understand this mechanism you should be able to write products for the following reactions:
 The addition products formed in reactions of alkenes with mercuricacetate and boron hydrides (compounds shown at the bottom of of thereagent list) are normally not isolated, but instead are converted toalcohols by a substitution reaction. These important synthetictransformations are illustrated for 2-methylpropene by the followingequations, in which the electrophilic moiety is colored red and thenucleophile blue. The top reaction sequence illustrates the oxymercuration procedure and the bottom is an example of hydroboration. The light blue vertical line separates the addition reaction on theleft from the substitution on the right. The atoms or groups that havebeen added to the original double bond are colored orange in the finalproduct. In both cases the overall reaction is the addition of water tothe double bond, but the regioselectivity is reversed. Theoxymercuration reaction gives the product predicted by Markovnikov'srule; hydroboration on the other hand gives the "anti-Markovnikov"product. Complementary reactions such as these are important becausethey allow us to direct a molecular transformation whichever way isdesired. Mercury and boron are removed from the organic substrate in the secondstep of oxymercuration and hydroboration respectively. These reactionsare seldom discussed in detail; however, it is worth noting that themercury moiety is reduced to ****llic mercury by borohydride (probablyby way of radical intermediates), and boron is oxidized to borate bythe alkaline peroxide. Addition of hydroperoxide anion to theelectrophilic borane generates a tetra-coordinate boron peroxide,having the general formula R3B-O-OH(-). This undergoes successiveintramolecular shifts of alkyl groups from boron to oxygen, accompaniedin each event by additional peroxide addition to electron deficientboron. The retention of configuration of the migrating alkyl group isattributed to the intramolecular nature of the rearrangement. Since the oxymercuration sequence gives the same hydration product as acid-catalyzed addition of water (see Brønsted acid addition),we might question why this two-step procedure is used at all. Thereason lies in the milder reaction conditions used for oxymercuration.The strong acid used for direct hydration may not be tolerated by otherfunctional groups, and in some cases may cause molecular rearrangement (see above). The addition of borane, BH3, requires additional comment. In pure formthis reagent is a dimeric gas B2H6, called diborane, but in ether orTHF solution it is dissociated into a solvent coordinated monomer,R2O-BH3. Although diborane itself does not react easily with alkenedouble bonds, H.C. Brown(Purdue, Nobel Prize 1979) discovered that the solvated monomer addsrapidly under mild conditions. Boron and hydrogen have rather similarelectronegativities, with hydrogen being slightly greater, so it is notlikely there is significant dipolar character to the B-H bond. Sinceboron is electron deficient (it does not have a valence ****l electronoctet) the reagent itself is a Lewis acid and can bond to thepi-electrons of a double bond by displacement of the ether moiety fromthe solvated monomer. As shown in the following equation, this bondingmight generate a dipolar intermediate consisting of anegatively-charged boron and a carbocation. Such a species would not bestable and would rearrange to a neutral product by the shift of ahydride to the carbocation center. Indeed, this hydride shift isbelieved to occur concurrently with the initial bonding to boron, asshown by the transition state drawn below the equation, so the discreteintermediate shown in the equation is not actually formed.Nevertheless, the carbocation stability rule cited above remains auseful way to predict the products from hydroboration reactions. You may correct the top equation by clicking the button on its right.Note that this addition is unique among those we have discussed, inthat it is a single-step process. Also, all three hydrogens in boraneare potentially reactive, so that the alkyl borane product from thefirst addition may serve as the hydroboration reagent for twoadditional alkene molecules.  To examine models of B2H6. and its dissociation in THF Stereoselectivity in Addition Reactions to Double Bonds To examine models of B2H6. and its dissociation in THF Stereoselectivity in Addition Reactions to Double Bonds
As illustrated in the drawing on the right, the pi-bond fixesthe carbon-carbon double bond in a planar configuration, and does notpermit free rotation about the double bond itself. We see then that addition reactions to this function might occur inthree different ways, depending on the relative orientation of theatoms or groups that add to the carbons of the double bond: (i) theymay bond from the same side, (ii) they may bond from opposite sides, or(iii) they may bond randomly from both sides. The first twopossibilities are examples of stereoselectivity, the first being termed syn-addition, and the second anti-addition.Since initial electrophilic attack on the double bond may occur equallywell from either side, it is in the second step (or stage) of thereaction (bonding of the nucleophile) that stereoselectivity may beimposed. If the two-step mechanism described above is correct, and if thecarbocation intermediate is sufficiently long-lived to freely-rotateabout the sigma-bond component of the original double bond, we wouldexpect to find random or non-stereoselective addition in the products.On the other hand, if the intermediate is short-lived and factors suchas steric hindrance or neighboring group interactions favor one side inthe second step, then stereoselectivity in product formation is likely.The following table summarizes the results obtained from many studies,the formula HX refers to all the strong Brønsted acids. Theinteresting differences in stereoselectivity noted here provide furtherinsight into the mechanisms of these addition reactions. ReagentH–XX2HO–XRS–ClHg(OAc)2BH3 Stereoselectivitymixedantiantiantiantisyn 1. Brønsted Acid Additions The stereoselectivity of Brønsted acid addition issensitive to experimental conditions such as temperature and reagentconcentration. The selectivity is often anti, but reports of synselectivity and non-selectivity are not uncommon. Of all the reagentsdiscussed here, these strong acid additions (E = H in the followingequation) come closest to proceeding by the proposed two-step mechanismin which a discrete carbocation intermediate is generated in the firststep. Such reactions are most prone to rearrangement when this isfavored by the alkene structure. 2. Addition Reactions Initiated by Electrophilic Halogen The halogens chlorine and bromine add rapidly to a wide variety ofalkenes without inducing the kinds of structural rearrangements notedfor strong acids (first example below). The stereoselectivity of theseadditions is strongly anti, as shown in many of the following examples. An important principle should be restated at this time. The alkenesshown here are all achiral, but the addition products have chiralcenters, and in many cases may exist as enantiomeric stereoisomers. Inthe absence of chiral catalysts or reagents, reactions of this kindwill always give racemic mixtures if the products are enantiomeric. Onthe other hand, if two chiral centers are formed in the addition thereaction will be diastereomer selective. This is clearly shown by theaddition of bromine to the isomeric 2-butenes. Anti-addition tocis-2-butene gives the racemic product, whereas anti-addition to thetrans-isomer gives the meso-diastereomer. We can account both for the high stereoselectivity and the lack ofrearrangement in these reactions by proposing a stabilizing interactionbetween the developing carbocation center and the electron rich halogenatom on the adjacent carbon. This interaction, which is depicted forbromine in the following equation, delocalizes the positive charge onthe intermediate and blocks halide ion attack from the syn-********. The stabilization provided by this halogen-carbocation bonding makesrearrangement unlikely, and in a few cases three-membered cyclichalonium cations have been isolated and identified as trueintermediates. A resonance description of such a bromonium ionintermediate is shown below. The positive charge is delocalized overall the atoms of the ring, but should be concentrated at the moresubstituted carbon (carbocation stability), and this is the site towhich the nucleophile will bond.  Becausethey proceed by way of polar ion-pair intermediates, chlorine andbromine addition reactions are faster in polar solvents than innon-polar solvents, such as hexane or carbon tetrachloride. However, inorder to prevent solvent nucleophiles from competing with the halideanion, these non-polar solvents are often selected for these reactions.In water or alcohol solution the nucleophilic solvent may open thebromonium ion intermediate to give an α-halo-alcohol or ether, togetherwith the expected vic-dihalide. Such reactions are sensitive to pH andother factors, so when these products are desired it is necessary tomodify the addition reagent. Aqueous chlorine exists as the followingequilibrium, Keq ≈ 10-4. By adding AgOH, the concentration of HOCl canbe greatly increased, and the chlorohydrin addition product obtainedfrom alkenes. Cl2 + H2O  HOCl + HCl The more widely used HOBr reagent, hypobromous acid, is commonly madeby hydrolysis of N-bromoacetamide, as shown below. Both HOCl and HOBradditions occur in an anti fashion, and with the regioselectivitypredicted by this mechanism (OH bonds to the more substituted carbon ofthe alkene). CH3CONHBr + H2O HOBr + CH3CONH2 Vicinal halohydrins provide an alternative route for the epoxidation of alkenes over that of reaction with peracids. As illustrated in the following diagram, a base induced intramolecular substitution reactionforms a three-membered cyclic ether called an epoxide. Both thehalohydrin formation and halide displacement reactions arestereospecific, so stereoisomerism in the alkene will be reflected inthe epoxide product ( i.e. trans-2-butene forms atrans-disubstituted epoxide). A general procedure for forming theseuseful compounds will be discussed in the next section.  [center] 3. Addition Reactions Involving Other Cyclic Onium Intermediates
 
Sulfenyl chloride additions are initiated by the attack of anelectrophilic sulfur species on the pi-electrons of the double bond.The resulting cationic intermediate may be stabilized by thenon-bonding valence ****l electrons on the sulfur in exactly the sameway the halogens exerted their influence. Indeed, a cyclic sulfoniumion intermediate analogous to the bromonium ion is believed to bestrepresent this intermediate (see drawing on the left).
Two advantages of the oxymercuration method of adding water to a doublebond are its high anti-stereoselectivity and the lack of rearrangementin sensitive cases. These characteristics are attributed to amercurinium ion intermediate, analogous to the bromonium ion discussedabove. In this case it must be d-orbital electrons that are involved inbonding to carbon. A drawing of this intermediate is shown on the right.
Hydroboration Stereoselectivity
The hydroboration reaction is among the few simple addition reactions that proceed cleanly in a syn fashion. As noted above,this is a single-step reaction. Since the bonding of the double bondcarbons to boron and hydrogen is concerted, it follows that the geometry of this addition must be syn.Furthermore, rearrangements are unlikely inasmuch as a discretecarbocation intermediate is never formed. These features areillustrated for the hydroboration of α-pinene in the followingequation. Since the hydroboration procedure is most commonly used tohydrate alkenes in an anti-Markovnikov fashion, we also need to knowthe stereoselectivity of the second oxidation reaction, whichsubstitutes a hydroxyl group for the boron atom. Independent study hasshown this reaction takes place with retention of configuration so the overall addition of water is also syn.

The hydroboration of α-pinene also provides a nice example of sterichindrance control in a chemical reaction. In the less complex alkenesused in earlier examples the plane of the double bond was often a planeof symmetry, and addition reagents could approach with equal ease fromeither side. In this case, one of the methyl groups bonded to C-6(colored blue in the equation) covers one face of the double bond,blocking any approach from that side. All reagents that add to thisdouble bond must therefore approach from the side opposite this methyl.
[center] 
Addition of hydrogen to a carbon-carbon double bond is called hydrogenation.The overall effect of such an addition is the reductive removal of thedouble bond functional group. Regioselectivity is not an issue, sincethe same group (a hydrogen atom) is bonded to each of the double bondcarbons. The simplest source of two hydrogen atoms is molecularhydrogen (H2), but mixing alkenes with hydrogen does not result in anydiscernible reaction. Although the overall hydrogenation reaction isexothermic, a high activation energy prevents it from taking placeunder normal conditions. This restriction may be circumvented by theuse of a catalyst, as shown in the following diagram.

Catalysts are substances that changes the rate (velocity) of a chemicalreaction without being consumed or appearing as part of the product.Catalysts act by lowering the activation energy of reactions, but theydo not change the relative potential energy of the reactants andproducts. Finely divided ****ls, such as platinum, palladium andnickel, are among the most widely used hydrogenation catalysts.Catalytic hydrogenation takes place in at least two stages, as depictedin the diagram. First, the alkene must be adsorbed on the surface ofthe catalyst along with some of the hydrogen. Next, two hydrogens shiftfrom the ****l surface to the carbons of the double bond, and theresulting saturated hydrocarbon, which is more weakly adsorbed, leavesthe catalyst surface. The exact nature and timing of the last events isnot well understood.
As shown in the energy diagram, the hydrogenation of alkenes isexothermic, and heat is released corresponding to the ΔE (coloredgreen) in the diagram. This heat of reaction can be used to evaluatethe thermodynamic stability of alkenes having different numbers ofalkyl substituents on the double bond. For example, the following tablelists the heats of hydrogenation for three C5H10 alkenes which give thesame alkane product (2-methylbutane). Since a large heat of reactionindicates a high energy reactant, these heats are inverselyproportional to the stabilities of the alkene isomers. To a roughapproximation, we see that each alkyl substituent on a double bondstabilizes this functional group by a bit more than 1 kcal/mole.
Alkene Isomer (CH3)2CHCH=CH2
3-methyl-1-butene CH2=C(CH3)CH2CH3
2-methyl-1-butene(CH3)2C=CHCH3
2-methyl-2-butene Heat of Reaction
( ΔHº ) –30.3 kcal/mole–28.5 kcal/mole–26.9 kcal/mole 
From the mechanism shown here we would expect the addition of hydrogento occur with syn-stereoselectivity. This is often true, but thehydrogenation catalysts may also cause isomerization of the double bondprior to hydrogen addition, in which case stereoselectivity may beuncertain.
The formation of transition ****l complexes with alkenes has beenconvincingly demonstrated by the isolation of stable platinum complexessuch as Zeise's salt, K[PtCl3(C2H4)].H2O, andethylenebis(triphenylphosphine)platinum, [(C6H5)3P]2Pt(H2C=CH2). In thelatter, platinum is three-coordinate and zero-valent, whereas Zeise'ssalt is a derivative of platinum(II). A model of Zeise's salt and adiscussion of the unusual bonding in such complexes may be viewed by clicking here.Similar complexes have been reported for nickel and palladium, ****lswhich also function as catalysts for alkene hydrogenation.
Anon-catalytic procedure for the syn-addition of hydrogen makes use ofthe unstable compound diimide, N2H2. This reagent must be freshlygenerated in the reaction system, usually by oxidation of hydrazine,and the strongly exothermic reaction is favored by the elimination ofnitrogen gas (a very stable compound). Diimide may exist as cis-transisomers; only the cis-isomer serves as a reducing agent. Examples ofalkene reductions by both procedures are shown on the right.
5. Oxidations
(i) Hydroxylation
Dihydroxylated products (glycols) are obtained by reaction withaqueous potassium permanganate (pH > 8) or osmium tetroxide inpyridine solution. Both reactions appear to proceed by the samemechanism (shown below); the ****llocyclic intermediate may be isolatedin the osmium reaction. In basic solution the purple permanganate anionis reduced to the green manganate ion, providing a nice color test forthe double bond functional group. From the mechanism shown here wewould expect syn-stereoselectivity in the bonding to oxygen, andregioselectivity is not an issue.
When viewed in context with the previously discussed additionreactions, the hydroxylation reaction might seem implausible.Permanganate and osmium tetroxide have similar configurations, in whichthe ****l atom occupies the center of a tetrahedral grouping ofnegatively charged oxygen atoms. How, then, would such a speciesinteract with the nucleophilic pi-electrons of a double bond? Apossible explanation is that an empty d-orbital of the electrophilic****l atom extends well beyond the surrounding oxygen atoms andinitiates electron transfer from the double bond to the ****l, in muchthe same fashion noted above for platinum. Back-bonding of thenucleophilic oxygens to the antibonding π*-orbital completes thisinteraction. The result is formation of a ****llocyclic intermediate,as shown below.

(ii) Epoxidation
Some oxidation reactions of alkenes give cyclic ethers in whichboth carbons of a double bond become bonded to the same oxygen atom.These products are called epoxides or oxiranes.An important method for preparing epoxides is by reaction withperacids, RCO3H. The oxygen-oxygen bond of such peroxide derivatives isnot only weak (ca. 35 kcal/mole), but in this case is polarized so thatthe acyloxy group is negative and the hydroxyl group is positive(recall that the acidity of water is about ten powers of ten weakerthan that of a carboxylic acid). If we assume electrophilic characterfor the OH moiety, the following equation may be written.

It is unlikely that a dipolar intermediate, as shown above, is actuallyformed. The epoxidation reaction is believed to occur in a single stepwith a transition state incorporating all of the bonding events shownin the equation. Consequently, epoxidations by peracids always havesyn-stereoselectivity, and seldom give structural rearrangement. Youmay see the transition state by clicking the Change Equation button.Presumably the electron shifts indicated by the blue arrows induce acharge separation that is immediately neutralized by the green arrowelectron shifts.
The previous few reactions have been classified as reductions or oxidations, depending on the change in oxidation state of the functional carbons.It is important to remember that whenever an atom or group is reduced,some other atom or group is oxidized, and a balanced equation mustbalance the electron gain in the reduced species with the electron lossin the oxidized moiety, as well as numbers and kinds of atoms. Startingfrom an alkene (drawn in the box), the following diagram shows ahydrogenation reaction on the left (the catalyst is not shown) and anepoxidation reaction on the right. Examine these reactions, and foreach identify which atoms are reduced and which are oxidized.

Epoxides may be cleaved by aqueous acid to give glycols that are often diastereomeric with those prepared by the syn-hydroxylation reactiondescribed above. Proton transfer from the acid catalyst generates theconjugate acid of the epoxide, which is attacked by nucleophiles suchas water in the same way that the cyclic bromonium ion described aboveundergoes reaction. The result is anti-hydroxylation of thedouble bond, in contrast to the syn-stereoselectivity of the earliermethod. In the following equation this procedure is illustrated for acis-disubstituted epoxide, which, of course, could be prepared from thecorresponding cis-alkene. This hydration of an epoxide does not changethe oxidation state of any atoms or groups.

(iii) Oxidative Cleavage of Double Bonds
Ozonolysis
In determining the structural formula of an alkene, it is oftennecessary to find the ******** of the double bond within a given carbonframework. One way of accomplishing this would be to selectively breakthe double bond and mark the carbon atoms that originally formed thatbond. For example, there are three isomeric alkenes that all give2-methylbutane on catalytic hydrogenation. These are 2-methyl-2-butene(compound A), 3-methyl-1-butene (compound B) and 2-methyl-1-butene(compound C), shown in the following diagram. If the double bond iscleaved and the fragments marked at the cleavage sites, the ******** ofthe double bond is clearly determined for each case. A reaction thataccomplishes this useful transformation is known. It is called ozonolysis, and its application to each of these examples may be seen by clicking the "Show Reaction" button.
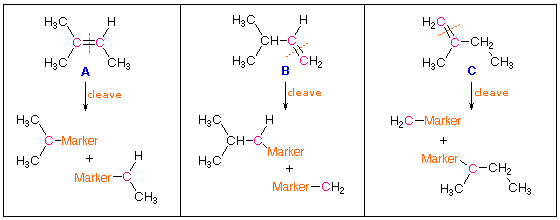
Ozone, O3, is an allotrope of oxygen that adds rapidly to carbon-carbondouble bonds. Since the overall change in ozonolysis is more complexthan a simple addition reaction, its mechanism has been extensivelystudied. Reactive intermediates called ozonides have been isolated fromthe interaction of ozone with alkenes, and these unstable compounds maybe converted to stable products by either a reductive workup (Zn dustin water or alcohol) or an oxidative workup (hydrogen peroxide). Theresults of an oxidative workup may be seen by clicking the "Show Reaction"button a second time. Continued clicking of this button repeats thecycle. The chief difference in these conditions is that reductiveworkup gives an aldehyde product when hydrogen is present on a doublebond carbon atom, whereas oxidative workup gives a carboxylic acid orcarbon dioxide in such cases. The following equations illustrateozonide formation, a process that is believed to involve initialsyn-addition of ozone, followed by rearrangement of the extremelyunstable molozonide addition product. They also show the decompositionof the final ozonide to carbonyl products by either a reductive oroxidative workup.

 Glycol Cleavage Glycol Cleavage
The vicinal glycols prepared by alkene hydroxylation (reaction withosmium tetroxide or permanganate) are cleaved to aldehydes and ketonesin high yield by the action of lead tetraacetate (Pb(OAc)4) or periodic acid(HIO4). This oxidative cleavage of a carbon-carbon single bond providesa two-step, high-yield alternative to ozonolysis, that is oftenpreferred for small scale work involving precious compounds. A generalequation for these oxidations is shown below. As a rule, cis-glycolsreact more rapidly than trans-glycols, and there is evidence for theintermediacy of heterocyclic intermediates (as shown), although theirformation is not necessary for reaction to occur.

Free Radical Reactions of Alkenes
1. Addition of Radicals to Alkenes
Protons and other electrophiles are not the only reactive speciesthat initiate addition reactions to carbon-carbon double bonds.Curiously, this first became evident as a result of conflicting reportsconcerning the regioselectivity of HBr additions. As noted earlier, theacid-induced addition of HBr to 1-butene gave predominantly2-bromobutane, the Markovnikov Ruleproduct. However, in some early experiments in which peroxidecontaminated reactants were used, 1-bromobutane was the chief product.Further study showed that an alternative radical chain-reaction,initiated by peroxides, was responsible for the anti-Markovnikovproduct. This is shown by the following equations.

This free radical chain addition competes very favorably with theslower ionic addition of HBr described earlier, especially in non-polarsolvents. It is important to note, however, that HBr is unique in thisrespect. The radical addition process is unfavorable for HCl and HIbecause one of the chain steps becomes endothermic (the second for HCl& the first for HI).
Other radical addition reactions to alkenes have been observed, oneexample being the peroxide induced addition of carbon tetrachlorideshown in the following equation
RCH=CH2 + CCl4 (peroxide initiator) [size=16]—> RCH ClCH2 CCl3 The best known and most important use of free radical addition to alkenes is probably polymerization.Since the addition of carbon radicals to double bonds is energeticallyfavorable, concentrated solutions of alkenes are prone toradical-initiated polymerization, as illustrated for propene by thefollowing equation. The blue colored R-group represents an initiatingradical species or a growing polymer chain; the propene monomers arecolored maroon. The addition always occurs so that the more stableradical intermediate is formed. RCH2(CH3)CH · + CH3CH=CH2 —> RCH2(CH3)CH- CH2(CH3)CH· + CH3CH=CH2 —> RCH2(CH3)CH CH2(CH3)CH-CH2(CH3)CH· —> etc. 2. Allylic Substitution
We noted earlier that benzylic and allylic sites are exceptionally reactive in free radical halogenation reactions.Since carbon-carbon double bonds add chlorine and bromine in liquidphase solutions, radical substitution reactions by these halogens areoften carried out at elevated temperature in the gas phase (firstequation below). Formation of the ionic π-complexes that areintermediates in halogen addition is unfavorable in the absence ofpolar solvents, and entropy generally favors substitution over addition.
The brominating reagent, N-bromosuccinimide (NBS), has proven usefulfor achieving allylic or benzylic substitution in CCl4 solution attemperatures below its boiling point (77 ºC). One suchapplication is shown in the second equation.

HBr + (CH2CO)2NBr —> Br2 + (CH2CO)2NH This mechanism is essentially the same as that for the free radical halogenationof alkanes, with NBS serving as a source of very low concentrations ofbromine. Unsymmetrical allylic radicals will react to give tworegioisomers. Thus, 1-octene on bromination with NBS yields a mixtureof 3-bromo-1-octene (ca. 18%) and 1-bromo-2-octene (82%) - both cis andtrans isomers.
RCH2CH=CH2 + (CH2CO)2NBr —> RCHBrCH=CH2 + RCH=CHCH2Br + (CH2CO)2NH
[/center] [/size][/center] [/center] | |
| | | | ابــن الاسلام
إدارة المنتدى
الجنس : السٌّمعَة : 101 الْمَشِارَكِات : 12744 النقاط/ : 22644 العـمــر : 34 الدولة : المتصفح :
| | موضوع: رد: Alkenes...... الثلاثاء نوفمبر 09, 2010 11:39 am | |
| Dienes
1. Properties of Dienes
When considering compounds having two or more double bonds inamolecule, it is useful to identify three distinct ways in whichthesefunctions may be oriented with respect to each other. First, thedoublebonds may be separated by one or more sp3-hybridized carbonatoms, asin 1,5-hexadiene. In this circumstance each double bondbehavesindependently of the other, and we refer to them as isolated.Asecond relationship has the double bonds connected to each other byasingle bond, as in 1,3-hexadiene, and we refer to this arrangement as conjugated.Finally,two double bonds might share a carbon atom, as in1,2-hexadiene. Thecentral carbon atom in such a system issp-hybridized, and we call suchdouble bonds cumulated. These three isomers are shown in the following diagram, and three other similar isomers will be displayed on clicking the Change Examples button. In cases where stereoisomers are possible only the E-isomer is shown.
Another stereoisomeric factor associated with conjugated dienes will be demonstrated by clicking the Change Examples button a second time. Rotation about the single bond joining the two double bonds (colored blue) converts a trans-like s-trans conformation to its s-cisform.The energy barrier to this conformational isomerization isnormally low,and the s-trans conformer is often more stable than thes-cis conformer,as shown in the diagram.

Thesecategories are based on more than obvious structural variations.We findsignificant differences in the chemical properties of dienesdependingon their structural type.  For example, catalytic hydrogenation converts all the dienes shown here to the alkane hexane, but the heats of reaction (heat of hydrogenation)reflectcharacteristic differences in their thermodynamic stability.This isillustrated in the diagram on the right. Taking the heatofhydrogenation of 1-hexene (30.1 kcal/mole) as a reference, we findthatthe isolated diene, 1,5-hexadiene, as expected, generates doublethisheat of reaction on conversion to hexane. The cumulateddiene,1,2-hexadiene, has a 6 kcal/mole higher heat of reaction,indicating itis less stable than the isolated diene by this magnitude.On the otherhand, conjugation of double bonds seems to stabilize adiene by about 5kcal/mole. The increase in stability of 2,4-hexadieneover1,3-hexadiene (both are conjugated) is due to the increased doublebondsubstitution of the former, a factor noted earlier for simple alkenes. For example, catalytic hydrogenation converts all the dienes shown here to the alkane hexane, but the heats of reaction (heat of hydrogenation)reflectcharacteristic differences in their thermodynamic stability.This isillustrated in the diagram on the right. Taking the heatofhydrogenation of 1-hexene (30.1 kcal/mole) as a reference, we findthatthe isolated diene, 1,5-hexadiene, as expected, generates doublethisheat of reaction on conversion to hexane. The cumulateddiene,1,2-hexadiene, has a 6 kcal/mole higher heat of reaction,indicating itis less stable than the isolated diene by this magnitude.On the otherhand, conjugation of double bonds seems to stabilize adiene by about 5kcal/mole. The increase in stability of 2,4-hexadieneover1,3-hexadiene (both are conjugated) is due to the increased doublebondsubstitution of the former, a factor noted earlier for simple alkenes.
The stabilization of dienes by conjugation is less dramatic than the aromatic stabilization of benzene.Nevertheless,similar resonance and molecular orbital descriptions ofconjugation maybe written. A resonance description, such as the oneshown here,involves charge separation, implying a relatively smalldegree ofstabilization.
CH2=CH-CH=CH2  (+)CH2-CH=CH-CH2:(–) (+)CH2-CH=CH-CH2:(–)
A molecular orbital model for 1,3-butadiene is shown below. Notethatthe lobes of the four p-orbital components in each pi-orbitalarecolored differently and carry a plus or minus sign. Thisdistinctionrefers to different phases, defined by the mathematical waveequationsfor such orbitals. Regions in which adjacent orbital lobesundergo aphase change are called nodes.Orbital electron densityis zero in such regions. Thus a singlep-orbital has a node at thenucleus, and all the pi-orbitals shown herehave a nodal plane that isdefined by the atoms of the diene. This isthe only nodal surface in thelowest energy pi-orbital, π1. Higherenergy pi-orbitals have anincreasing number of nodes.

To examine a model of the p-orbital components for the s-cis conformer of a 1,3-diene.
To examine the molecular orbitals of both s-trans and s-cis-1,3-butadiene
2. Addition Reactions of Dienes
Addition reactions of isolated dienes proceed more or less asexpectedfrom the behavior of simple alkenes. Thus, if one molar*****alent of1,5-hexadiene is treated with one *****alent of bromine amixture of5,6-dibromo-1-hexene, 1,2,5,6-tetrabromohexane and unreacteddiene isobtained, with the dibromo compound being the major product(about 50%).
CH2=CH(CH2)2CH=CH2 + Br2 BrCH2CHBr(CH2)2CH=CH2 + BrCH2CHBr(CH2)2CHBrCH2Br + CH2=CH(CH2)2CH=CH2
5,6-dibromo-1-hexene1,2,5,6-tetrabromohexane1,5-hexadiene
Similarreactions of conjugated dienes, on the other hand, often giveunexpectedproducts. The addition of bromine to 1,3-butadiene is anexample. Asshown below, a roughly 50:50 mixture of3,4-dibromo-1-butene (theexpected product) and 1,4-dibromo-2-butene(chiefly the E-isomer) isobtained. The latter compound is remarkablein that the remaining doublebond is found in a ******** where therewas no double bond in thereactant. This interesting re********requires an explanation.
CH2=CH-CH=CH2 + Br2 BrCH2CHBr-CH=CH2 + BrCH2CH=CHCH2Br
3,4-dibromo-1-butene1,4-dibromo-2-butene
The expected addition product from reactions of this kind is the result of 1,2-addition, i.e. bonding to the adjacent carbons of a double bond. The unexpected product comes from 1,4-addition,i.e.bonding at the terminal carbon atoms of a conjugated diene with ashiftof the remaining double bond to the 2,3-********. These numbersrefer tothe four carbons of the conjugated diene and are not IUPACnomenclaturenumbers. Product compositions are often temperaturedependent, as theaddition of HBr to 1,3-butadiene demonstrates.
CH2=CH-CH=CH2 + HBr
reaction temperature CH3CHBr-CH=CH2 +
1,2 addition yield CH3CH=CHCH2Br
1,4 addition yield 0 ºC
40 ºC70%
15%30%
85%
Bondingof an electrophilic atom or group to one of the end carbonatoms of aconjugated diene (#1) generates an allyl cationintermediate. Suchcations are stabilized by charge delocalization, andit is thisdelocalization that accounts for the 1,4-addition productproduced insuch addition reactions. As shown in the diagram, thepositive charge isdistributed over carbons #2 and #4 so it is at thesesites that thenucleophilic component bonds. Note that resonancestabilization of theallyl cation is greater than comparable stabilization of 1,3-butadiene, because charge is delocalized in the former, but created and separated in the latter.
[center]  Anexplanation for the temperature influence is shown in thefollowingenergy diagram for the addition of HBr to 1,3-butadiene. Theinitialstep in which a proton bonds to carbon #1 is the rate determining step, as indicated by the large activation energy (light gray arrow). The second faster step is the product determining step,andthere are two reaction paths (colored blue for 1,2-addition andmagentafor 1,4-addition). The 1,2-addition has a smaller activationenergy than1,4-addition, but the 1,4-product is more stable than the1,2-product.At low temperatures, the products are formed irreversiblyand reflectthe relative rates of the two competing reactions. This istermed kinetic control. At higher temperatures, equilibrium is established between the products, and the thermodynamically favored 1,4-product dominates.

The unique character of conjugated dienes manifests itself dramatically in the Diels-Alder Cycloaddition Reaction.Acycloaddition reaction is the concerted bonding together oftwoindependent pi-electron systems to form a new ring of atoms. Whenthisoccurs, two pi-bonds are converted to two sigma-bonds, thesimplestexample being the hypothetical combination of two ethenemolecules togive cyclobutane. This does not occur under normalconditions, but thecycloaddition of 1,3-butadiene to cyanoethene(acrylonitrile) does, andthis is an example of the Diels-Alderreaction. The following diagramillustrates two cycloadditions, andintroduces several terms that areuseful in discussing reactions of thiskind.

The Diels-Alder reaction is animportant andwidely used method for making six-membered rings, as shownon theright. The reactants used in such reactions are a conjugateddiene,simply referred to as the diene, and a double or triple bond coreactant called the dienophile,becauseit combines with (has an affinity for) the diene. TheDiels-Aldercycloaddition is classified as a [4+2] process because thediene hasfour pi-electrons that shift position in the reaction and thedienophilehas two.
The Diels-Alder reaction is a single stepprocess, so thediene component must adopt a cis-like conformation inorder for the endcarbon atoms (#1 & #4) to bond simultaneously tothe dienophile.Such conformations are called s-cis, the sreferring tothe single bond connecting the two double bonds. The s-cisand s-transconformers of 1,3-butadiene are shown in the precedingdiagram. For manyacyclic dienes the s-trans conformer is more stablethan the s-cisconformer (due to steric crowding of the end groups),but the two aregenerally in rapid equilibrium, permitting the use ofall but the mosthindered dienes as reactants in Diels-Alder reactions.In its usualform, the diene component is electron rich, and the bestdienophiles areelectron poor due to electron withdrawing substituentssuch as CN, C=O& NO2. The initial bonding interaction reflectsthis electronimbalance, with the two new sigma-bonds being formedsimultaneously, butnot necessarily at equal rates.
Stereospecificity
We noted earlier that addition reactions of alkenes often exhibited stereoselectivity,inthat the reagent elements in some cases added syn and in othercasesanti to the the plane of the double bond. Both reactants intheDiels-Alder reaction may demonstrate stereoisomerism, and when theydoit is found that the relative configurations of the reactantsarepreserved in the product (the adduct). The followingdrawingillustrates this fact for the reaction of 1,3-butadienewith(E)-dicyanoethene. The trans relationship of the cyano groups inthedienophile is preserved in the six-membered ring of theadduct.Likewise, if the terminal carbons of the diene bearsubstituents, theirrelative configuration will be retained in theadduct. Using theearlier terminology, we could say that bonding to boththe diene andthe dienophile is syn. An alternative description,however, refers tothe planar nature of both reactants and terms thebonding in each caseto be suprafacial (i.e. to or from the same face of each plane). This stereospecificity also confirms the synchronous nature of the 1,4-bonding that takes place.

(i) The reaction always creates a new six-membered ring. When intramolecular, another ring may also be formed.
(ii) The diene component must be able to assume a s-cis conformation.
(iii) Electron withdrawing groups on the dienophile facilitate reaction.
(iv) Electron donating groups on the diene facilitate reaction.
(v) Steric hindrance at the bonding sites may inhibit or prevent reaction.
(vi) The reaction is stereospecific with respect to substituent configuration in both the dienophile and the diene.
Thesefeatures are illustrated by the following eight examples, one ofwhichdoes not give a Diels-Alder cycloaddition. Try to predict thecourse ofeach reaction before disclosing the answers by pressing the "Show Products" button. The formation of a new six-membered ring should be apparent in every case where reaction occurs.

There is no reaction in example D because this diene cannot adopt a s-cis orientation. In examples B, C, F, G & Hatleast one of the reactants is cyclic so that the product has morethanone ring, but the newly formed ring is always six-membered. Inexample Bthe the same cyclic compound acts as both the dienecolored blue) andthe dienophile (colored red). The adduct has threerings, two of whichare the five-membered rings present in thereactant, and the third isthe new six-membered ring (shaded lightyellow). Example C hasan alkyne as a dienophile (colored red),so the adduct retains a doublebond at that ********. This double bondcould still serve as adienophile, but in the present case the diene issufficiently hinderedto retard a second cycloaddition. The quinonedienophile in reaction Fhas two dienophilic double bonds.However, the double bond with twomethyl substituents is less reactivethan the unsubstituted dienophiledue in part to the electron donatingproperties of the methyl groups andin part to steric hindrance. Thestereospecificity of the Diels-Alderreaction is demonstrated byexamples A, E & H. In A & H the stereogenic centers lie on the dienophile, whereas in E these centers are on the diene. In all cases the configuration of the reactant is preserved in the adduct.
Cyclic dienes, such as those in examples B, C & G, give bridged bicyclicadductsfor which an additional configurational feature must bedesignated. Asshown in the following diagram, there are two possibleconfigurationsfor compounds of this kind. If a substituent (coloredmagenta here) isoriented cis to the longest or more unsaturated bridge(colored bluehere), it is said to be endo. When directed trans to the bridge it is exo.Whenthe Diels-Alder reaction forms bridged bicyclic adducts andanunsaturated substituent is located on this bicyclic structure (as in B & G), the chief product is normally the endo isomer "Alder's Endo Rule". Example C does not merit such a nomenclature, since stereoisomeric orientations of the substituent are not possible.

[/center] | |
| | | | جزائري أصيل
المدير العام


الجنس : السٌّمعَة : 5 الْمَشِارَكِات : 4460 النقاط/ : 4544 العـمــر : 42 الدولة :
| | موضوع: رد: Alkenes...... الجمعة أغسطس 26, 2011 6:49 pm | |
| عمل المعروف يدوم
و الجميل دائما محفوظ
لا تفكروا في يوم أنسى
أنكم وقفتم مع طلاب العلوم
عجزت الكلمات تعبر
عن مدى الجميل و العرفان
الذي بدر منكم تجاه طلاب غرداية
كل الجميل للعمل الذي
ما أظن ينساه إنسان
فبارك الله فيكم
وفي عملكم الموزون
دمتم بطيب النسيم
وعبق الرحيق المختوم
شكرا لكم | |
| | | | | | Alkenes...... | |
|
| | صلاحيات هذا المنتدى: | لاتستطيع الرد على المواضيع في هذا المنتدى
| |
| |
| |
|







